个性化增材制造医疗器械注册技术审查指导原则(5)
(十三)产品的临床评价要求
个性化增材制造医疗器械临床评价的目标是为了获得安全性和有效性数据,评价个性化增材制造医疗器械在治疗特殊病例和特殊解剖部位疾患的作用。如通过临床试验评价产品安全性和有效性,临床试验应当符合《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局中华人民共和国国家卫生和计划生育委员会令第25号)的相应要求,临床试验机构应当按要求在国家药品监督管理局备案。
1. 无可替代产品情形
病源有限或标准化产品不适宜作为对照的,可以开展不少于10例的观察研究,每个临床机构应当开展不少于5例研究。可以和申请人以往的历史数据进行综合分析,符合本指导原则要求的可以纳入统计。属于临床急需或罕见病情况的可以依据相关规定进行试验。
应当注意对个性化增材制造产品特定安全性和有效性指标进行观察。例如个性化医疗器械使用过程中发生的不良事件、使用过程中临床医生操作性能、植入假体的初始稳定性、患者的功能恢复及生存质量的早期改善等。
根据疾病类型和临床获益确定研究终点,研究终点为至少3个月,但该临床病例应当给予持续跟踪,直至临床转归的稳定状态。
2. 需要进行同类对照产品情形
如可设立阳性对照,则应当参照随机、平行、对照的前瞻性临床试验原则,进行非劣效性临床试验。
2.1入选、排除标准
对于需要进行临床试验的个性化增材制造医疗器械,其受试者应当严格遵从患者获益的前提,从需要进行个性化医疗器械治疗的患者人群中选出。申请人及临床试验机构应当根据申报产品的设计特征及其适用范围制定其临床试验的入选/排除/退出标准,不符合所有入选标准或者符合任何一项排除标准的研究对象应被排除。
2.2受试者退出标准及退出受试者的处理
2.2.1退出标准
①受试者撤回知情同意书;
②严重违反验证方案;
③研究者认为不再适合继续进行临床验证者;
④在临床验证期间妊娠的妇女;
⑤受试者死亡;
⑥受试者失访;
⑦申办者要求终止验证。
2.2.2退出受试者的处理
①最后一次生命体征记录、术后情况和局部体征检查资料、影像学检查资料,记录合并用药和不良事件等;
②将终止验证的时间和原因详细记录在病例报告表上;
③对因不良事件而终止验证的病人必须随访至不良事件得到解决或稳定。
④《医疗器械临床试验质量管理规范》规定的其他相关要求。
2.3个性化医疗器械植入手术操作执行要求
为降低手术植入环节的风险,应当针对不同部位的个性化医疗器械应用,建立手术操作的文本及图示规范指导实施。根据需要选择计算机导航和辅助导板进行精确手术,以确保个性化医疗器械的精准安装。
2.4临床试验持续时间与窗口期
临床试验的持续时间取决于安全性和有效性数据的获取,针对个性化3D打印器械的孔隙结构利于骨长入形成远期稳定的特点,临床试验可重点考量器械的初始稳定性,临床试验持续时间至少3个月。随访内容包括患者主诉、体格检查、影像评价、功能评估等。
2.5临床试验评价指标及判定标准
对于需进行临床试验的个性化医疗器械,根据植入部位不同,参考现有常规产品或根据病变部位特点设立主要评价指标和次要评价指标,并明确评估方法。主要评价指标是与试验目的有本质联系的、能确切反映器械疗效或安全性的指标。次要评价指标是与试验目的相关的辅助性指标。
2.6对照产品的选择
对开展临床试验的个性化医疗器械,对照产品应当尽可能选择目前临床正广泛使用的、对相应适应症的疗效已被证实并得到公认的等效产品。对照产品的材料、设计、适应症与试验产品具有可比性,应当提供对照产品的选择依据。
2.7统计分析方法
应当明示具体的统计分析方法以及统计分析软件及版本。数据分析时应当考虑数据的完整性,所有签署知情同意并使用了受试产品的受试者必须纳入分析。数据的剔除或偏移数据的处理必须有科学依据和详细说明。
临床试验的数据分析应当基于不同的分析集,通常包括全分析集(Full Analysis Set,FAS)、符合方案集(Per Protocol Set,PPS)和安全集(Safety Set,SS),研究方案中应当明确各分析集的定义。全分析集中脱落病例,其主要研究终点的缺失值的填补方法等应当在方案中事先予以说明,并进行不同分析策略的灵敏度分析,以评价缺失数据对研究结果稳定性的影响。
主要研究终点指标的分析应当同时在全分析集和符合方案集上进行,安全性指标的分析应当基于安全集。
临床试验数据的分析应当采用国内外公认的经典统计分析方法。临床试验方案应当明确统计检验的类型、检验假设、判定疗效有临床意义的界值(非劣效界值)等,界值的确定应当有依据。
对于主要研究终点,统计结果需采用点估计及相应的95%可信区间进行评价。不能仅将p值作为对主要研究终点进行评价的依据。
对验证期间发生的所有有害事件的种类、严重程度、发生频率及与验证产品的关系将列表描述。
申请人应当提供基于所有临床试验数据的统计分析报告,以便临床试验组长单位根据此报告撰写临床试验总结报告。
3.个性化医疗器械根据其内在规律,可以采用临床评价、动物实验和功能试验等方法,进行综合风险评估。临床评价可以依据风险要素进行设定,评估对风险要素的控制程度。
(十四)产品的不良事件历史记录
应当按要求收集、记录、提交产品相关的不良事件历史记录。
(十五)产品说明书和标签要求
产品说明书、标签和包装标识应当符合《医疗器械说明书和标签管理规定》要求,还应当符合相关国家标准、行业标准的要求,例如YY/T 0466.1―2016《医疗器械 用于医疗器械标签、标记和提供信息的符号 第1部分:通用要求》。
除已批准信息外,说明书和标签中应当明确产品为个性化医疗器械,补充患者特征标识、临床医生书面确认产品设计方案的信息或文件编号及其他需要补充的信息。
三、审查关注点
应用本指导原则进行技术审评时,除审查用于骨、关节和口腔硬组织的无源植入性医疗器械产品的安全性和有效性外,还重点关注实现个性化产品设计、完成增材制造加工的能力和质量。
四、编写单位
本指导原则由国家药品监督管理局医疗器械技术审评中心编写并负责解释。
(责任编辑:admin)

 MGA医疗联盟:以系统化思
MGA医疗联盟:以系统化思 国家统计局:中国3D打印设
国家统计局:中国3D打印设 ICC将制定混凝土3D打印墙
ICC将制定混凝土3D打印墙 三项增材制造金属粉末国家
三项增材制造金属粉末国家 工信部等新规:工业级3D打
工信部等新规:工业级3D打 2027年产值突破1500亿元(
2027年产值突破1500亿元( 洞察2023:全球药
洞察2023:全球药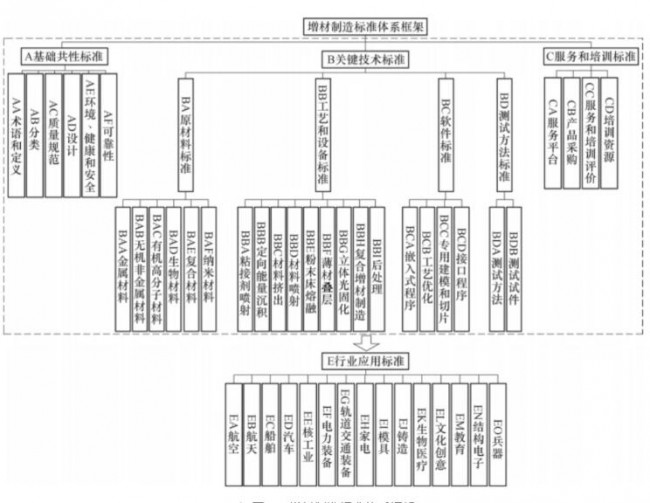 3D打印-增材制造
3D打印-增材制造 《智能制造知识体
《智能制造知识体 2021年中国及31省
2021年中国及31省 全球工程标准协会
全球工程标准协会 毛孔的SLM金属3D
毛孔的SLM金属3D

